
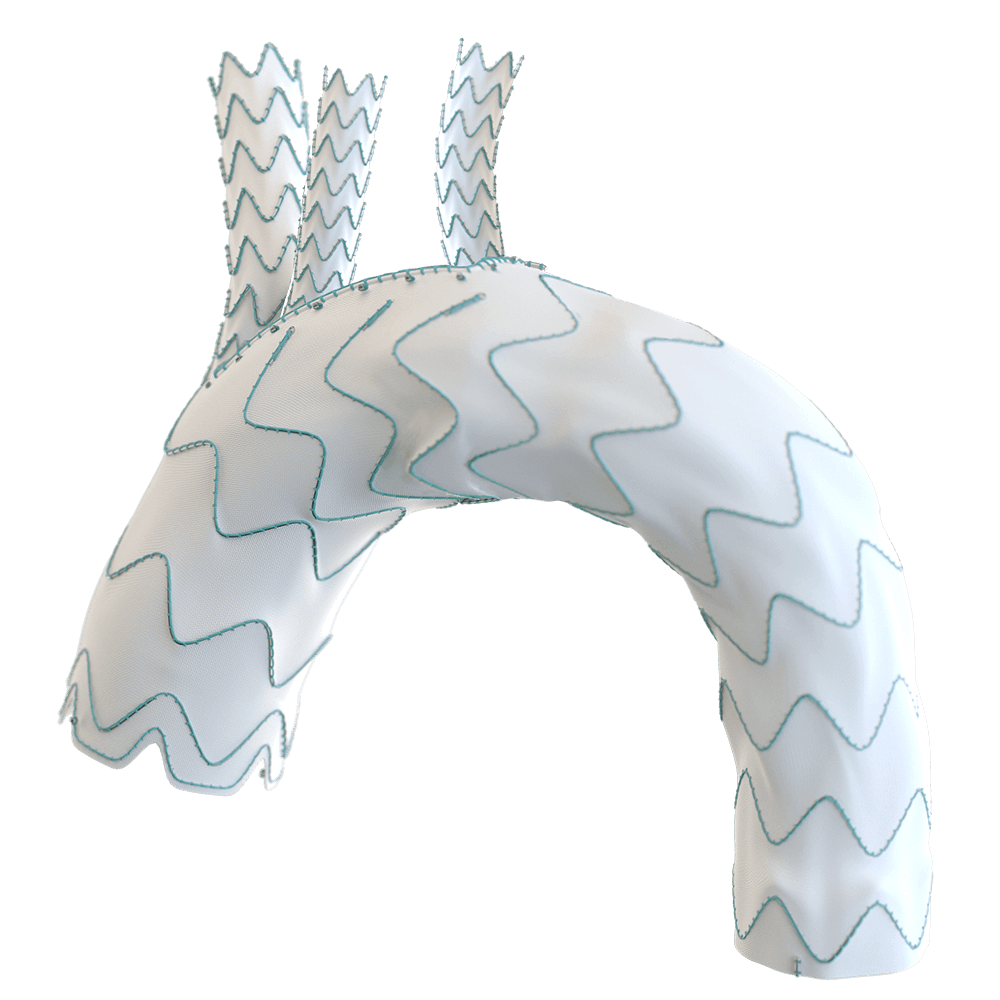
The RelayBranch Thoracic Stent-Graft System is implanted in patients with thoracic aortic arch pathologies requiring treatment that includes coverage of the innominate and left common carotid arteries.
The purpose of the FDA’s Breakthrough Device Designation program is to fast-track the regulatory review process for certain medical technologies and device-led combination products that satisfy certain criteria; these include providing a more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions. The aim of the program is to provide patients and healthcare professionals with timely access to important breakthrough medical devices.
The RelayBranch Thoracic Stent-Graft System consists of a main body graft which is deployed in the ascending aorta and features two anterograde tunnels that give way to a large cannulation window. Branch grafts are then deployed within these tunnels for both the innominate artery and left common carotid artery. The RelayBranch Thoracic Stent-Graft System is part of a feasibility study currently enrolling in the United States.
Wilson Szeto, MD, Chief of Cardiovascular Surgery, Penn Presbyterian University of Pennsylvania Health System, Philadelphia, commented: “This breakthrough designation from the FDA will allow US physicians to treat patients with a less invasive treatment option through the innovative technology of the RelayBranch endovascular device. This is a treatment for patients who are high risk for open surgical repair with no alternative therapies available.”
Megan Indeglia, Global Vice President of Regulatory Affairs at Terumo Aortic added: “This announcement from the FDA is very encouraging and we look forward to continued collaboration with the regulatory authorities to advance treatment options for patients with complex aortic pathology.”
The RelayBranch Stent-Graft System complements Terumo Aortic’s innovative portfolio of surgical, endovascular and hybrid devices to treat every segment of the aorta.


